
Search the Community
Showing results for tags 'schizophrenia'.
Found 8 results
-

An Interview with The Deputy Director for Safety at the FDA's Division of Psychiatry Products - WOW!!
FeralCatman posted a topic in In the media
This link is to an interview with The Deputy Director for Safety at the FDA's Division of Psychiatry Products. The FDA's stance is that they determine efficacy but safety is subjective and is largely up to doctor and patient to determine for themselves based on individual circumstances. He went on to state that it is up to the patient to thoroughly research each drug for themselves and that informed consent is basically up to the patient. That pretty much says it all. It also goes on to discuss what is lacking in the studies and the FDA's official response is that their data on withdrawal and long term effects is seriously lacking and needs improvement but that it is very hard to do all of that which is why it doesn't get done. It also states that the drug companies tend to prefer a certain level of ambiguity when it comes to safety. WTH??? 🤬 https://www.theinnercompass.org/blog/are-psychiatric-medications-safe-fdas-answer-may-surprise-you -
 Not sure I'm posting this in the right place -- am following the logic that an academic dissertation falls in the research category. "Postpsychiatry's Challenge to the Chemical Treatment of Mental Distress" is psychologist Olga Runciman's 2013 master's thesis from the Department of Psychology at the University of Copenhagen. https://psycovery.com/images/Postpsychiatrys-challenge-Olga-Runciman-2013.pdf The link above is copied directly from psychologist Olga Runciman's professional website, as is the presentation below (I've made a few edits to the latter for clarity, fixed typo, etc.): Why Postpsychiatry Postpsychiatry does not seek to find solutions within psychiatry. Instead they advocate we should be moving beyond psychiatry, encouraging an acceptance that not all human problems can be explained within an illness paradigm. Postpsychiatry questions the paradigm of recovery which in psychiatry is often viewed from a perspective of a reduction or lessening of intensity of symptoms. Instead they promote supporting that it is the person herself who defines his recovery. I am critical of the medical model and psychiatric medication aligning myself with critical and postpsychiatry. For an in-depth look at why, you are welcome to download my thesis which describes this. Seven people labeled schizophrenic participated in qualitative interviews, giving voice to their experience of taking psychiatric medication. Three accept medication, two doubt its efficacy, and two are off medication completely. The voices of the interviewees introduce a world of context, meaning and understanding and show that madness is multifaceted and complex but understandable. The narratives of their medication experiences challenge the current psychiatric belief system. Link to clinical psychologist and psychotherapist Olga Runciman's professional website, psycovery, where I found all of the above: https://psycovery.com/index.php/en/about-psycovery
Not sure I'm posting this in the right place -- am following the logic that an academic dissertation falls in the research category. "Postpsychiatry's Challenge to the Chemical Treatment of Mental Distress" is psychologist Olga Runciman's 2013 master's thesis from the Department of Psychology at the University of Copenhagen. https://psycovery.com/images/Postpsychiatrys-challenge-Olga-Runciman-2013.pdf The link above is copied directly from psychologist Olga Runciman's professional website, as is the presentation below (I've made a few edits to the latter for clarity, fixed typo, etc.): Why Postpsychiatry Postpsychiatry does not seek to find solutions within psychiatry. Instead they advocate we should be moving beyond psychiatry, encouraging an acceptance that not all human problems can be explained within an illness paradigm. Postpsychiatry questions the paradigm of recovery which in psychiatry is often viewed from a perspective of a reduction or lessening of intensity of symptoms. Instead they promote supporting that it is the person herself who defines his recovery. I am critical of the medical model and psychiatric medication aligning myself with critical and postpsychiatry. For an in-depth look at why, you are welcome to download my thesis which describes this. Seven people labeled schizophrenic participated in qualitative interviews, giving voice to their experience of taking psychiatric medication. Three accept medication, two doubt its efficacy, and two are off medication completely. The voices of the interviewees introduce a world of context, meaning and understanding and show that madness is multifaceted and complex but understandable. The narratives of their medication experiences challenge the current psychiatric belief system. Link to clinical psychologist and psychotherapist Olga Runciman's professional website, psycovery, where I found all of the above: https://psycovery.com/index.php/en/about-psycovery -
Hello everyone, I don’t even know where to start. My 19 years old girlfriend was diagnosed with paranoid schizophrenia, depression and OCD in 2015 when she was 13 years old. She was also hospitalized 3 times. We’re together for 9 months. This month, roughly month after tapering off Cipralex, my girlfriend started having, which I think are withdrawal symptoms (suicidal ideation, really bad anhedonia, drowsiness, sometimes pressure like feeling on the whole head). She told me she experienced anhedonia even in year 2018 after mental “downfall” (complete fall to the depression and suicidal black hole) after cold turkeying Zoloft and Abilify (don’t remember the dosage, haven’t taken those regularly) and feelings haven’t come back since, maybe only briefly. Before that she normally felt every emotion. She had psychiatric appointment yesterday and she’s supposed to start taking Abilify again, which I don’t know it’s a good idea with the cocktail she already have. Her psychiatrist also said, there’s possibility of her not taking these drugs the whole lifetime and also said the tapering Cipralex wasn’t that fast while simultaneously starting Brintellix (vortioxetine). I just want her to be happy and help. I don’t know how to help her with getting her feelings back and whether that’s still a WD symptom after 2018 fall. The thing she’s deeply sad about the most is the emotional numbness. She’s also experiencing loss of libido from year 2018. I have a hope she doesn’t have to take those drugs forever, but don’t know whether she should start tapering them now or not or maybe later? Is Abilify a good idea? Is emotional numbness caused by the pills or rather withdrawal? Any advice on what to do is greatly appreciated. Thank you bunch, everyone.
-

☼ The loving, positive voice hearer turned 'ZombieMode' by antipsychotics
ZombieMode posted a topic in Introductions and updates
Hi All, Diagnosed schizophrenic here currently on a 300mg depot injection of Abilify monthly until March 2016, on a community treatment order. Am doing everything I can to get off medication ASAP, as am experiencing akathisia, anhedonia, complete loss of libido, numbness, and a loss of spirituality. I'm sure most of you know how horrible this can be, especially when previously I was a healthy & happy, loving guy full of life and energy. In total I'll be on the abilify for a year, what are my chances of recovery? Has anyone in a similar position ever come off medication and found their voices to return (mine were extremely loving, and quite special to me)? Could anyone provide me with tips to detox/cleanse, or peace of mind that I will recover? I smoke ciggarettes, take st johns wort, to try to counter the effects of the abilify, which help, along with numerous other supplements. Exercise regularly, eat as healthy as possible. Thanks for reading, I really appreciate any help or guidance on the topic of antipsychotics. -
Hello all! I feel very happy to have found this place today! I am a 49 year old guy, single, not because of lack of women but because of the difficulty to keep a relation besides of being chemically castrated by psychiatric medications for 35 years in a row. Before dealing with my mental issues I was a very healthy guy, loved sports, had excellent grades and was very sociable and happy. I come from a very dysfunctional family with an alcoholic father and a sex offender neighbor who abused me many times but I never told anyone. At that time of my life I could not realize that I had been traumatized by the sexual assaults of my neighbor, but because of a change in behavior, my mother knew something was going on with me. So she decided to take me to a Pediatric Psychiatrist and the Pandora's box jut opened. I was a living trauma and for the first time in my life I was medicated. It has been 35 years without having a normal, enjoyable life My whole health decayed big time in many aspects. During treatments I developed: diabetes, hypercholesterol, hypertriglyceridemia, cardiovascular disease, whole body nerve damage, fibromyalgia, darkening and peeling of the skin in my legs, burning tongue syndrome, impotence and harsh tooth decay. I could be a 49 year old man but specialist doctors tell me that I am a high risk patient and that the situation of my physical health is the same as of an 85 year old man and that with all my health conditions, I could die anytime. It wasn't til last week that I decided to put an end to all of this, no matter how. I have used any single psychotropic medication, the old and the new and all of them are really bad for you but the FDA and the Big Pharma Industry want your money and will try you to keep on consuming their products. They don' really care if you feel good or not at all. I was feeling miserable, lying on bed for weeks because my body pains were so severe that I could not barely walk. Then, one day I started a research on psychiatric medications side effects in detail and I could relate to my state of physical health. So I made another research on how to withdraw from psychiatric medications but using natural supplements and that information has helped me big time. My current cocktail last week was Depakote 2,000mg Seroquel 800mg Efexxor 225mg Xanax 6mg Ambien 10mg Adderall XR 60mg Neurontin 1,600mg Estazolam 4mg I made a research of every single one, selected the hardest to eliminate, and decreased their dosages. Right now I am taking: Depakote 200mg Seroquel 200mg Efexxor 75mg Xanax 4mg Ambien 10mg Adderall XR 30mg Neurontin and Estazolam are history. It feels like kind of hell but this is my goal and I know I can accomplish it and at the end I will have the kind of life that I deserve. For next week I will keep on reducing dosages and going into "full time " natural supplementation with: Vitamin D, Chelated Magnesium, Turmeric, L-Carnitine, Vitamin E, Fish Oil, Vitamin C 1,000, Garlic, Ginckgo Biloba, Coenzime Q10, Flavonoids, Copper, Alpha LipoicAcid, B Complex and B-12 separately. Do your research for your own good. You were not born with all those meds and undesirable side effects, there is a light at the end of the tunnel! The information I posted is based on my own opinions and experiences and in NO way I instruct you to do the same, I just think that my history may inspire you in making things the better for you. Many blessings to all, HEMARO
-

Chouinard, 2017 Antipsychotic-Induced Dopamine Supersensitivity Psychosis: Pharmacology, Criteria, and Therapy
DoctorMussyWasHere posted a topic in From journals and scientific sources
This paper is one of the most thorough I have come across in relation to its topic: Supersensitivity Psychosis, the reason antipsychotics are in fact pro-psychotics. Antipsychotic-Induced Dopamine Supersensitivity Psychosis: Pharmacology, Criteria, and Therapy Chouinard G.a,b,g · Samaha A.-N.c, d · Chouinard V.-A.e, f · Peretti C.-S.g · Kanahara N.h · Takase M.i· Iyo M.h, i It's long and thorough - over an hour at 200wds/pm The lead author, Guy Chouinard, is notable for his role in identifying this phenomenon in 1978. This blog post has a little more background on Chouinard. Although the content may be a little on the technical side, which is say more of interest to laboratory researchers, the paper proposes diagnostic guidelines. Whether or not these will make it into future manuals such as the DSM and ICD remains to be seen, however it would be interesting to see how this would sit alongside modern descriptions of disorders, many of which have absorbed the side effects of medications into their lists of symptoms, thereby masking them, and leading to ever-increasing doses. I think the descriptions of what is drug-induced versus what is a latent problem may potentially be useful. The fact that it recognises a distinction and attempts to define it at all is fairly noteworthy. I've included that section, preceded by the abstract, and links to the original content. This comes to around 6000 words - about 30 minutes reading. Abstract The first-line treatment for psychotic disorders remains antipsychotic drugs with receptor antagonist properties at D2-like dopamine receptors. However, long-term administration of antipsychotics can upregulate D2 receptors and produce receptor supersensitivity manifested by behavioral supersensitivity to dopamine stimulation in animals, and movement disorders and supersensitivity psychosis (SP) in patients. Antipsychotic-induced SP was first described as the emergence of psychotic symptoms with tardive dyskinesia (TD) and a fall in prolactin levels following drug discontinuation. In the era of first-generation antipsychotics, 4 clinical features characterized drug-induced SP: rapid relapse after drug discontinuation/dose reduction/switch of antipsychotics, tolerance to previously observed therapeutic effects, co-occurring TD, and psychotic exacerbation by life stressors. We review 3 recent studies on the prevalence rates of SP, and the link to treatment resistance and psychotic relapse in the era of second-generation antipsychotics (risperidone, paliperidone, perospirone, and long-acting injectable risperidone, olanzapine, quetiapine, and aripiprazole). These studies show that the prevalence rates of SP remain high in schizophrenia (30%) and higher (70%) in treatment-resistant schizophrenia. We then present neurobehavioral findings on antipsychotic-induced supersensitivity to dopamine from animal studies. Next, we propose criteria for SP, which describe psychotic symptoms and co-occurring movement disorders more precisely. Detection of mild/borderline drug-induced movement disorders permits early recognition of overblockade of D2 receptors, responsible for SP and TD. Finally, we describe 3 antipsychotic withdrawal syndromes, similar to those seen with other CNS drugs, and we propose approaches to treat, potentially prevent, or temporarily manage SP. Introduction Pharmacological Mechanisms by Which Antipsychotics Potentially Increase D2 Receptor Density and/or Function Clinical Characteristics and Consequences of SP in the Era of SGAs Evolving Conceptual Framework of Antipsychotic-Induced Dopamine SP in Line with Other CNS Drug Withdrawal Syndromes Pharmacological Link between a Compensatory Increase in D2/D2High Receptor Density and SP Guidelines for SP and TD Prevention Choice of the Antipsychotic Guidelines for SP Treatment Guidelines for the Management of Antipsychotic Withdrawal Evolving Conceptual Framework of Antipsychotic-Induced Dopamine SP in Line with Other CNS Drug Withdrawal Syndromes Classification of Antipsychotic Withdrawal Syndromes Based on ongoing research into SP characteristics, along with evidence for withdrawal symptoms found with other CNS drug classes, we present a conceptual framework [1,41] for identifying SP and other antipsychotic withdrawal syndromes (Tables 1, 2). Chouinard and colleagues [1,31,89,90] reported 3 types of withdrawal syndromes associated with CNS drugs, including opiates, barbiturates, and alcohol: (1) new or classical withdrawal symptoms; (2) rebound; and (3) postwithdrawal disorders (Table 1). These 3 types of withdrawal syndromes need to be differentiated from relapse and recurrence of the original illness. Relapse is defined as the gradual return to symptoms seen before treatment and entails a return of the ongoing episode, while recurrence is defined as a new episode of the illness (Table 1). Table 1 Type of antipsychotic withdrawal compared to relapse and recurrence [41] Table 2 Diagnostic criteria for new symptoms induced by antipsychotic treatment New and rebound symptoms occur up to 6 weeks after drug discontinuation/dose reduction/switch, depending on the drug terminal plasma half-life (t1/2). New and rebound symptoms of CNS drugs have been reported to be more frequent and severe with short t1/2 and high-potency drugs [1,31,89,90]. New and rebound withdrawal symptoms are usually of short duration, persisting for a few hours to 2 weeks with complete recovery. However, if symptoms persist for more than 6 weeks, they become part of postwithdrawal disorders [41]. Fava et al. [91] proposed to replace the term discontinuation syndrome often used for selective serotonin reuptake inhibitors (SSRIs)/selective noradrenaline reuptake inhibitors (SNRIs) [92] and antipsychotic medications [93] by “withdrawal.” CNS withdrawal occurs when the pharmacological effects of a drug diminish during medication discontinuation/dose reduction/switch, or in-between doses, which indicates an underlying pharmacological mechanism. The term discontinuation refers to the medical act of drug discontinuation or patient self-discontinuation. Furthermore, the term discontinuation is misleading since withdrawal symptoms may also occur with dose reduction or in-between doses for rapid-onset short-acting drugs. Examples include alprazolam and lorazepam, which can induce clock watching in-between doses, and clozapine which can induce late afternoon psychosis when given as a single dose at bedtime [41]. We will give criteria for each of the 3 types of withdrawal associated with antipsychotics [1,41,94]. New Symptoms New symptoms are the classic symptoms of withdrawal. They are not part of the patient's illness. New symptoms can be minor (anxiety, insomnia, and tremor) or major (seizures, psychosis, and death) [95]. Some are common to all CNS drugs, including narcotics, barbiturates, and alcohol, but each class of CNS drugs has its own specific new symptoms [1,41,90]. New symptoms common to CNS drugs include: nausea, headache, tremor, sleep disturbances, decreased concentration, anxiety, irritability, agitation, aggression, depression, or dysphoria [41], as listed in Tables 3 and 4. On the other hand, new drug-specific symptoms are linked to receptor binding affinities, and result from receptor supersensitivity which leads to increased neurotransmission due to removal of receptor blockade by antipsychotic treatment when medication is discontinued or switched, or dose is reduced [1,41,90]. New and rebound symptoms occurring during withdrawal and switch from and to SGAs (aripiprazole, asenapine, iloperidone, lurasidone, olanzapine, paliperidone, quetiapine, risperidone, sertindole, ziprasidone, and amisulpride) were reviewed recently by Cerovecki et al. [94]. In Tables 1 and 2, we integrate some of their findings on treatment-emergent symptoms related to removal of blockade or stimulation of dopamine and other receptors (serotonin, muscarinic, adrenergic, and histaminic receptors) by SGAs [94], and divide new antipsychotic-specific symptoms into: (1) serotonin withdrawal syndrome (equivalent to serotonin syndrome): flu-like symptoms, diarrhea, disorientation, coma, hyperreflexia, and stimulus-inducible clonus; (2) muscarinic withdrawal syndrome (also called cholinergic rebound syndrome): nausea, vomiting, abdominal cramps, hypothermia, increased salivation, tremor, parkinsonism, restlessness, and insomnia; (3) adrenergic withdrawal syndrome: increased blood pressure, headache, anxiety, agitation, increased heart rate, myocardial infarction, angina pectoris, palpitation, chest pain, presyncope, tremulousness, sweating, increased heart rate, myocardial infarction, hyperthermia, and fear; and (4) histaminic withdrawal syndrome: insomnia, agitation, tremulousness, increased seizure susceptibility, and amnesia (Table 2). Some new symptoms (Table 2) have been reported to occur more than 6 weeks after discontinuation: myocardial infarction and death after discontinuation of β-blockers [96]; hyperreflexia and inducible clonus after abrupt discontinuation of clozapine [97]; inducible seizure after discontinuation of chronic H1antihistamine treatment in rodents [98], and persistent insomnia following drug discontinuation/dose reduction of central muscarinic anticholinergics [1,90], and alcohol [95]. Table 3 Diagnostic criteria for rebound psychosis induced by antipsychotic treatment Table 4 Diagnostic criteria for persistent postwithdrawal supersensitivity psychosis induced by antipsychotic treatment Rebound Rebound Syndromes Associated with CNS Drugs The second type of CNS drug withdrawal consists of rebound symptoms (Tables 1, 2) [41]. It refers to a sudden return of patient symptoms above pretreatment levels and is usually short lasting. Rebound symptoms should be distinguished from pharmacological rebound, which produces both new and rebound symptoms. Pharmacological rebound is caused by a sudden increase or decrease in receptor-mediated effects due to removal of receptor blockade following drug discontinuation/dose reduction. This includes adrenergic rebound [99], cholinergic rebound [100,101], serotonin rebound [97], and histamine rebound [98]. We make a distinction between new and rebound symptoms, though they may have the same pharmacological mechanism [1,41,94]. The difference between new and rebound symptoms is whether present before treatment and known to the patient (rebound) or not present before treatment (new) and known to the patient. Cholinergic rebound produces both new symptoms: nausea, vomiting, salivation, abdominal cramp, diarrhea, hypothermia, tremor, insomnia, depression, and rebound symptoms listed in Tables 1 and 2; adrenergic rebound produces increase or “overshoot” in blood pressure and in heart rate above pretreatment and is also associated with new symptoms such as insomnia and anxiety [99,102]. Rebound anxiety and insomnia, seen with short-acting benzodiazepines [1], are also associated with new symptoms [103]. Withdrawal insomnia [104] and rebound insomnia [105] were identified as separate entities in all-night EEG recording studies. Recently, we reviewed CNS drug rebound syndromes [41]. Several CNS drug rebound syndromes have been reported to occur following drug discontinuation after short-term use: REM rebound with barbiturates and non-benzodiazepine hypnotics [106,107]; rebound myocardial angina and infarction with propranolol [108]; rebound insomnia [105], rebound anxiety [103], and rebound panic [109], with benzodiazepines with short and intermediate t1/2(triazolam, lorazepam, and alprazolam); and rebound depression [1,41]. Rebound depression has also been observed at a greater frequency with high-potency (paroxetine) and/or short-acting SSRIs and SNRIs [1,41]. In 2 double-blind placebo studies, patients with major depression had their maintenance paroxetine, sertraline, or fluoxetine treatment interrupted with placebo substitution and treatment reinstitution [110,111]. In one of the studies [110], patients taking paroxetine and sertraline had a sudden worsening of depressive symptoms upon drug discontinuation with a return to baseline depression measurements after reinstitution of the treatment. In both studies [110,111], patients taking fluoxetine did not have rebound depression, confirming that fluoxetine with a long t1/2 including norfluoxetine is less likely to cause rebound [41]. In contrast, patients treated with paroxetine had rebound depression in both studies [41,110,111]. The prevalence of high-dose use [112] is greater among patients taking benzodiazepines with short-intermediate t1/2 (e.g., triazolam, lorazepam, and alprazolam) than patients taking benzodiazepines (clonazepam, clobazam, and diazepam) with long t1/2; these differences persist even after 12-year long-term use [112]. Rebound syndromes respond rapidly to reinstitution of the offending drug, which often leads patients to false beliefs about efficacy and need for the drug [90]. The psychological belief of needing the drug for fear of having symptoms return appears the same for short-acting and short t1/2 CNS drugs, including antipsychotics (quetiapine) [1], paroxetine (SSRI) [41], lorazepam [103], alprazolam [112], cocaine [113], and heroin [114]. Rapid-onset drugs with short t1/2 will have an increased risk of rebound upon withdrawal, as illustrated by cocaine and heroin [114]. Rebound symptoms can be persistent in some patients, for example long-lasting insomnia after withdrawal from sustained alcohol abstinence [95] and rebound psychosis after long-term antipsychotic treatment [1,14]. Rebound Psychosis The proposed diagnostic criteria for rebound psychosis are given in Table 3. Rebound psychosis is defined by a rapid return above pretreatment levels of at least one positive symptom listed in the Rating Scale for Psychotic Symptoms (RSPS) [73,74]. Rebound psychosis lasts up to 6 weeks after peak onset (Table 1); if it persists longer, it becomes a postwithdrawal disorder, as for new symptoms persisting more than 6 weeks. Rebound psychosis is generally short lasting and considered as a reversible form of SP, equivalent to reversible withdrawal TD [1]. Any rebound symptom, such as an increase in blood pressure, myocardial infarction, insomnia, or psychosis, has the potential to become a persistent postwithdrawal disorder when lasting more than 6 weeks, often lasting several months and even years [1,41,95,96]. As with other CNS drugs (benzodiazepine, SSRI/SNRI, heroin, and cocaine), antipsychotics that are rapidly eliminated and/or that rapidly dissociate from the D2 receptor including clozapine (time to peak concentrations in plasma, tmax: 3-4 h; t1/2: 16 h [115]) and quetiapine [116] (tmax: 1-1.8 h; t1/2: 7 h [115]) have greater risks of rebound when medication is reduced, switched [59], or discontinued [1,33,39,40]. Patients on clozapine monotherapy taken as a single dose at bedtime report late afternoon return of symptoms [1]. For both the immediate-release formulation (IR) and the extended-release formulation (XR) of quetiapine, it is common in clinical practice to have difficulty in decreasing the drug dose, as it causes rebound anxiety and rebound insomnia (the drug possessing anxiolytic and hypnotic effects [117]), and later causes an in-between dose clock watching for return of symptoms, e.g., patient watches the clock to make sure the next dose is not missed for fear that symptoms might return, when given once a day above 150 mg [117] over a 2-month period [1,41]. The pharmacokinetics [118] and the central D2 receptor occupancy pharmacodynamics [119] of IR quetiapine given twice daily and XR quetiapine given once daily are similar [118,119]. Rebound psychosis with SGAs was first seen with clozapine [33] and then quetiapine monotherapy [39], but was later seen with all SGAs [34,35,36], with overt prevalence depending on drug t1/2 and potency. Quetiapine and clozapine have a particular D2 pharmacological profile compared to other SGAs: loose binding displaced rapidly from the D2 receptor [120], fast dissociation from the D2receptor [116], brief and weak action of clozapine on brain dopamine systems [116,121], and high transient initial D2 occupancy by quetiapine with minimal occupancy at the end of the dose interval [122]. This D2 profile of clozapine and quetiapine leads to differences in SP manifestations [1]: elevated incidence of rebound psychosis with clozapine [1,33,39,101], which is a reversible withdrawal subtype of SP [1], and severe drug tolerance with IR quetiapine given as antipsychotic monotherapy [39]. This might be explained by initially high D2 occupancy followed rapidly by low occupancy at the end of the dosing interval [122]. Postwithdrawal Disorders Postwithdrawal Disorders of CNS Drugs Postwithdrawal disorders have been described with all classes of CNS drugs [1,41]. Early recognition of postwithdrawal disorders for a given class of drugs is facilitated by the presence of high-potency drugs combined with short t1/2 [1,123,124]. Due to the short duration of action, there is a rapid decrease in occupancy of receptors produced in greater number by potent blocking drugs, thus resulting in a greater number of receptors in their unblocked state [1,108,123,124]; this was the case for β-adrenergic blocking drugs. As early as 1975, dose was highlighted as an additional factor for early recognition of postwithdrawal disorder; 160-320 mg propranolol/day would produce an adrenergic rebound unmasked rapidly by discontinuation, which led to an increase in cardiac contractibility, heart rate, myocardial ischemia, and rapid death, even after short-term use (6-12 weeks) [108]. Interestingly, in this first report of adrenergic rebound, major complications such as death were seen in patients who responded better to propranolol [108], which is also a characteristic of SP, most often seen in good antipsychotic drug responders [14,72]. Following abrupt withdrawal (even gradual withdrawal) of β-adrenergic drugs after long-term use, it was relatively easy to demonstrate an overshoot adrenergic rebound with pretreatment symptoms reappearing suddenly - increase in blood pressure, heart rate, myocardial infarction, headache, and anxiety above pretreatment levels [96,102]. The early recognition of supersensitivity rebound with β-adrenergic drugs helped to identify its underlying receptor supersensitivity mechanism through catecholamine stimulation by isoprenaline (isoproterenol), a nonselective β-adrenergic receptor agonist in patients [99,124,125], and later identify persistent postwithdrawal disorders consisting of an increased risk of myocardial infarction and mortality following their discontinuation [96]. Houston and Hodge [102], in their review of β-adrenergic blocker withdrawal syndromes, described 7 studies showing rebound and persistent withdrawal with 80 mg/day propranolol and a study showing these withdrawal syndromes after only 7 days of propranolol use. Doses below 80 mg propranolol/day appear safe to use in psychiatric clinical practice, but gradual dose reduction of low-dose propranolol is also recommended especially in patients at risks of myocardial infarction and essential hypertension. Rebound anxiety and insomnia were also first demonstrated with short-acting and potent benzodiazepines [90,103,105], and later as postwithdrawal disorders for alcohol [95] and benzodiazepines [104,126]. Other known postwithdrawal disorders are major depression and dysphoria in cocaine [113,114] and amphetamine withdrawal [114]. Within a drug class, specific drugs such as fluphenazine, perphenazine, quetiapine, clozapine, triazolam, and paroxetine permitted identification of new postwithdrawal disorders [1,33,39,41,109,127]. For SSRIs and SNRIs, several postwithdrawal disorders after long-term use have been described; interestingly, these postwithdrawal disorders consist of disorders that can be treated successfully by SSRIs and SNRIs, such as obsessive-compulsive, panic, generalized anxiety, and gambling disorders [1,41,109,127,128,129]. All these postwithdrawal disorders have been shown to occur more frequently with paroxetine [41,109,129,130]. This suggests that paroxetine should not be used anymore. For antipsychotics, two persistent postwithdrawal disorders, TD and SP, have been described [14,28,30,31,131]. When the symptoms last less than 6 weeks, they are considered as withdrawal dyskinesia and rebound, or withdrawal psychosis, respectively. When the symptoms last longer, they become postwithdrawal disorders, TD, or SP [1]. Discontinuation of short- and long-term β-blocker therapy can uncover long-lasting β-adrenergic supersensitivity [96,99]. Similarly, it has been known since the 1970s that long- and short-term dopamine receptor blockade by antipsychotics can both produce receptor supersensitivity [132]. The persistence of the dopamine supersensitivity syndrome depends on the duration of the preceding blockade [132] and on the specific antipsychotics used (fluphenazine, perphenazine, clozapine, and quetiapine) [1,41]. Similarly to β-adrenergic supersensitivity which may occur early and severely with β-blockers [108], TD can occur after short-term use (1 month) of chlorpromazine (FGA) in a patient with schizophrenia [133], and in a nonpsychiatric patient after a 4-month treatment with a gastric preparation containing trifluoperazine (FGA) and an anticholinergic drug [134] in an irreversible and extremely severe form. Supersensitivity Psychosis The proposed diagnostic criteria [14] for SP as a CNS postwithdrawal disorder [41] are presented in Table 4. The criteria take into account results from the 3 recent studies in patients treated with SGAs (n = 505) reviewed above [34,35,36], which examine the clinical characteristics of SP, 2 specifically focusing on TR schizophrenia (online suppl. Table 2). In addition, we took into account the results from the 2 studies by Fallon and Dursun [37 ]and Fallon et al. [38], which examined the link between SP and relapse in treatment-adherent schizophrenic patients without drug discontinuation who were not taking clozapine or quetiapine [37,38]. Exacerbation of psychosis by dopamine partial agonists was added as a risk factor (minor criterion) following the study by Takase et al. [36]. Vulnerability to minor life events was explicitly added to the minor criterion of exacerbation of psychosis by stress, following the results of Fallon and Dursun [37 ]and Fallon et al. [38]. Finally, the criteria define severe therapeutic drug tolerance more precisely as a nonresponse to doses equivalent to ≥600 mg chlorpromazine/day/12 mg haloperidol/day, when the patient has previously responded to lower doses. In addition, compared to 1991 criteria, major criteria define positive symptoms specifically [73,74]. Major criteria C2, C3, or C5 do not need minor criteria as was shown by the SP criteria used in the 3 recent Japanese studies of SP (n = 505), which included both patients with and without TR schizophrenia (online suppl. Table 2) [34,35,36], and in the 2 relapse studies [37,38]. Major criteria define rapid psychotic exacerbation or relapse upon drug discontinuation/dose reduction/switch of antipsychotics, and drug tolerance which refers to the lessening of drug therapeutic effects with continued treatment and the need for increasing doses to achieve the same beneficial effect. Minor criteria are risk factors for SP (Table 4); they help to identify patients at risk for SP relapse. One minor criterion is needed if only one of the major criteria present is C1, C2, or C6 (Table 4). We will now describe how SP criteria might be linked to an increase in D2/D2High receptor density. Pharmacological Link between a Compensatory Increase in D2/D2High Receptor Density and SP All antipsychotics, including the most recent SGAs [26], have the potential to produce D2upregulation. For instance, using laboratory animals, Tarazi et al. [135] reported upregulation of D2 receptors by olanzapine, risperidone, and quetiapine. Kusumi et al. [136] showed an increase in striatal D2 density following subchronic treatment with doses of chlorpromazine and perospirone (SGA) that produce therapeutic levels of striatal D2 receptor occupancy but not with low doses of chlorpromazine, risperidone, and olanzapine. While some antipsychotics may not elevate the density of D2 receptors, they can still raise the number of dopamine D2High receptors [25]. The increase in the number of D2 and/or D2High receptors enhances dopamine-mediated transmission [4] (Fig. 1). This may lead to more severe psychosis than seen in previous episodes during SP relapse or exacerbations, with new schizophrenic symptoms of greater severity such as disorganized behavior [14]. Patients with SP are more vulnerable to stress [14,69], minor life events, and daily hassles [14,37,38,69]. Stress increases dopamine levels in the brain [137]. In the presence of increased D2/D2High density, stress-induced dopamine in the extracellular space will produce greater D2-mediated signaling, leading to vulnerability to minor life events in patients with SP [14,69] even without drug discontinuation/dose reduction [37,38]. SP patients without drug discontinuation/dose reduction could be considered as having an overt form of SP [1,14]. As dopamine binds to D2receptors in competition with antipsychotic drugs, antipsychotics with less D2 receptor affinity allow more dopamine to bind to the receptors, and psychotic relapses due to SP will appear more rapidly. Another feature of SP is the gradual tolerance to the therapeutic effect of antipsychotic medications, where previously efficacious doses can no longer adequately control psychotic symptoms. Continuous D2 occupancy by antipsychotics, within or above the threshold for prolactin and movement disorders (i.e., in the 72-78% range [4,138]), increases D2 density as a compensatory reaction to reduced dopamine-mediated signaling. As D2 density increases, the therapeutic level of D2 occupancy (65%) [4,138] becomes higher [4], and previously efficacious doses are insufficient to suppress or cover psychotic symptoms. In order to improve psychotic symptoms, at least temporarily, higher doses are required [20], and therapeutic drug tolerance further develops. This has also been shown in animal models of antipsychotic-like efficacy, where continuous blockade of D2 receptors by antipsychotics, at clinically representative levels, produces tolerance to ongoing treatment, and higher doses can temporarily restore efficacy [9,10]. The optimal range of D2 occupancy in drug-naïve schizophrenia has been established [138], and theoretically determines the optimal dose range for antipsychotic drugs [4]. Therefore, patients who previously responded well to antipsychotics, but now have become poor responder or have histories of maintenance treatment with doses equivalent or higher than 600 mg chlorpromazine/day and 12 mg haloperidol/day are at increased risk for SP. Poor response to 2 antipsychotics at doses equivalent to or higher than 600 mg chlorpromazine/day is a criterion for TR schizophrenia [75,76]. Following antipsychotic discontinuation or sudden dose reduction, psychotic relapse develops more rapidly in patients with SP than in those without [69]. As the antipsychotic drug is eliminated with its specific t1/2, endogenous dopamine would now have access to a greater number of D2 and D2High receptors (Fig. 1), explaining why psychotic symptoms appear more rapidly in SP patients during drug discontinuation/dose reduction/switch of medication. However, the delay before the appearance of psychosis will also depend on the t1/2 of each drug. As optimal therapeutic D2 occupancy increases [4], increasing the antipsychotic dose would further mask TD [139] and SP [1], produce parkinsonism and negative symptoms, but also temporarily improve positive symptoms [4,34]. Some patients with severe SP who are also taking high doses of antipsychotics may exhibit all of these symptoms at the same time [1,34,140]. This mixed syndrome of drug-induced movement disorders and psychosis is temporally improved by giving the antipsychotic in divided doses [69]. Thus, SP may be masked, overt, or mixed [1], like TD [19]. The fact that SP can be overt during dose reduction, and masked by the causing drug during treatment [14,28,30], explains the therapeutic tolerance observed when antipsychotics can no longer mask psychotic symptoms at previously efficacious doses, and further increases in doses are necessary to obtain similar therapeutic effects [14,28,30]. Tolerance needs to be identified early to avoid increasing doses. Once therapeutic tolerance is identified, instead of dose increases to cover SP, strategies that we propose here should be considered, primarily the use of low doses of antiseizure drugs [1], the use of the minimal therapeutic dose of antipsychotic, and regular but intermittent dosing. Switching from one antipsychotic to another can potentially induce or uncover SP [14,37,69,94]. Switching antipsychotics will allow SP to emerge in patients either with or without a past history of SP [36]. This suggests that a medication switch can unmask SP induced by previous antipsychotic treatment, particularly in patients taking doses equivalent to ≥600 mg chlorpromazine/day/12 mg haloperidol/day. SP often occurs in the presence or history of drug-induced movement disorders, most often parkinsonism or TD [14,28,30,37]. This is expected since parkinsonism and other drug-induced movements are the best predictors for TD [1,71,81] and SP [1,37,38]. As striatal D2 occupancy by antipsychotics exceeds 65%, a clinical response is likely to appear, hyperprolactinemia when >72% and parkinsonism and movement disorders when >78% [138]. The therapeutic window of 65-72% D2 occupancy proposed by Kapur et al. [138] in their PET study of first-episode schizophrenia is relatively narrow and corresponds to doses >0.5 mg haloperidol/day [138]. Iyo et al. [4] suggested to extend the therapeutic window to >78%, i.e., when movement disorders appear, since movement disorders are easier to detect clinically and also because not all antipsychotics increase prolactin. Therefore, continuous D2 occupancy by antipsychotics above the narrow therapeutic window of 65-72% [138] or above the 72-78% threshold for prolactin/movement disorders may produce undetected parkinsonism and SP [1,4,19,28]. As already mentioned, Kapur et al. [138] found that the threshold level of striatal D2 receptor occupancy for movement disorders is >78% in first-episode schizophrenia. While catalepsy is seen in rats with doses that occupy >80% [141,142], dopamine supersensitivity can be seen at doses that occupy <80% [9,10,11,12]. Indeed, in animal models of dopamine supersensitivity, the doses we have studied occupy 73-74% of striatal D2receptors [9,10,11,12]. Table 5 Guidelines for the prevention of supersensitivity psychosis (SP) and tardive dyskinesia (TD) The second step (Table 5) is the administration of minimal therapeutic doses during maintenance treatment following the acute phase. The dose given in the acute phase is reduced gradually when initiating the maintenance treatment [148]. During the acute phase of psychosis, excessive dopamine release competes with antipsychotics for D2 receptor binding and depletes dopamine in the synaptic cleft [149], which creates a delay in reaching a new equilibrium. The third step (Table 5) is to keep D2 occupancy within the optimal therapeutic range while maintaining the minimal therapeutic dose. This must take into account the side effects and patient's tolerability, which could be predicted by 4 pharmacokinetic parameters (clearance, volume of distribution, t1/2, and bioavailability) [150]. In clinical practice, one should take into account that peak-to-trough plasma concentrations vary greatly and will depend on a number of variables. These include dosing interval and drug formulation [151], D2 receptor affinities, lipophilicity, active metabolites, patient characteristics (ethnicity and gender), concomitant medications, comorbid diseases, and general physical health [151,152]. While it is generally agreed that for oral SGAs (risperidone, olanzapine, clozapine, quetiapine, and ziprasidone) pharmacokinetic parameters are not sufficient for selection [115], selection of SGAs should be based on efficacy, side effects/tolerability, drug discontinuation-induced withdrawal symptoms and postwithdrawal disorders [1], and drug formulations [4,151]. For LA injectable antipsychotics, pharmacokinetic data in product monographs are of limited value for clinicians to choose the interval of administration [153]. In their recent review, Lee et al. [153] recommend using t1/2 for LA injectable SGAs to decide on the dosing interval in clinical practice. Continuous blockade of D2 receptors could contribute to the emergence of SP according to research conducted in animals. This work shows that when using the same achieved dose, peak level of D2 receptor occupancy, route, and duration of treatment, continuous antipsychotic treatment/D2 receptor blockade (as achieved via subcutaneous osmotic minipump) favors dopamine supersensitivity, while intermittent treatment/D2 receptor blockade (via daily subcutaneous injection) does not [9,11,154]. There are reports that intermittent antipsychotic administration via daily subcutaneous injection can produce dopamine supersensitivity [155,156,157,158]. However, the antipsychotic doses used are quite large and would produce excessively high and clinically unrepresentative levels of striatal D2 receptor blockade [159]. Continuous antipsychotic treatment could promote dopamine supersensitivity because it disrupts dopaminergic signaling unremittingly. This evokes compensatory changes including an increase in the number of striatal D2 and D2High receptors in the striatum (Fig. 1) [9,10,50]. In contrast, intermittent antipsychotic treatment allows at least some level of dopaminergic signaling to occur in between peaks in D2 receptor occupancy. This might be sufficient to prevent the brain changes that produce dopamine supersensitivity. The challenge is how to define “intermittent treatment” in clinical research and clinical practice. The clinician will often use short interdosing intervals (e.g., not taking antipsychotics on the weekends), to achieve minimal therapeutic dose [1]. In patients, there is likely a “break point” where if the antipsychotic dose/striatal D2 receptor occupancy has been low enough for long enough, the treatment might not be efficacious anymore to prevent relapses, thus being below the minimum therapeutic dosing. However, continuous dosing is not always necessary to maintain therapeutic efficacy. One prospective study by McCreadie et al. [160] indicates that intermittent administration with short interdosing intervals (as modeled in the animal studies described above [9,11,154]) can be as effective as continuous dosing in preventing psychotic relapse. In this double-blind study, patients were randomly assigned to either continue treatment with fluphenazine decanoate given every 2 weeks (continuous treatment, n = 18) or be switched to daily pimozide equivalent given 4 times a week (intermittent treatment, n = 16) [160]. Relapse rates over the next 9 months were similar in both groups. A second prospective study was a 6-month, double-blind, controlled trial in outpatients with schizophrenia stabilized for ≥3 months on oral antipsychotic given once daily [161]. The patients were randomly assigned to either treatment as usual (n = 18) or the same daily dose given orally every other day (n = 17; risperidone = 6, olanzapine = 11). Over the 6-month interval, symptom exacerbation and number of hospital admissions were similar in both groups. Thus, the 17 patients who had a 50% reduction in their maintenance dose given as alternate-day treatment (the “extended dosing” group) had similar relapse rates compared to those who kept taking their treatment as usual once a day [161]. However, reduction in the maintenance dose on one hand and continuous versus intermittent treatment on the other were confounded. To directly evaluate the effects of continuous versus intermittent antipsychotic treatment, a study would have to give the same “equivalent” dose daily versus every other day (e.g., 2 mg risperidone/day once daily versus 4 mg risperidone every other day). “Extended dosing” by giving treatment every other day is one strategy to reduce antipsychotic dose by 50%. The authors did not mention the length of prior antipsychotic treatment or the prior daily milligram dose of the 3 medications taken (risperidone, olanzapine, and loxapine). It is likely that the D2 occupancy was lower than the narrow therapeutic window of 65-72% found by Kapur et al. [138] for the acute/subacute phase and for first-episode schizophrenia. The McCreadie study [160] is more interesting in this respect since patients received a daily dose of oral pimozide 4 times a week (Monday to Thursday; intermittent dosing strategy), which was equivalent to the previous maintenance dose of LA injectable fluphenazine decanoate; intermittent continuous dosing strategy), thus testing the concept of intermittent antipsychotic treatment to achieve minimum therapeutic dosing and prevent dopamine supersensitivity. Similarly, Tsuboi et al. [162] conducted a single-blind, randomized, prospective, 1-year study including patients treated with oral olanzapine (n = 34) and oral risperidone (n = 34). Patients were stabilized for 6 months before entering the study. Patients were randomly assigned to receive either a treatment regimen producing continuous blockade of D2 >65% or a treatment regimen producing noncontinuous receptor blockade >65%. The continuous D2 blockade group had an estimated D2 occupancy >65% at trough and the noncontinuous blockade group had an estimated peak level of D2 occupancy >65% with an estimated trough level of <65%. The noncontinuous group had target doses of 7.1 mg olanzapine/day and 1.9 mg risperidone/day versus 10.4 and 3.4 mg/day for the continuous group, respectively, the latter producing >65% of estimated D2 occupancy. The authors did not find differences between both treatment regimens regarding the prevention of psychotic relapses. Again, here we have low- versus higher-dose maintenance treatment. However, the dosing interval of both drugs is not mentioned, and concomitant medications are not reported. The only information is “choice of dose at the discretion of the subject's physician on record.” Finally, we do not think that the study by Tsuboi et al. [162] is strictly a comparison of continuous versus noncontinuous but also low- versus high-dose treatment. In addition, a PET study by Nyberg et al. [163] showed that continuous D2 receptor blockade >65% was not necessary to prevent psychotic relapse. The study included 9 schizophrenic outpatients stabilized on haloperidol decanoate given intramuscularly every 4 weeks [163]. Despite an average fall of 52% in D2 receptor occupancy at the end of the injection interval compared to the occupancy level 1 week after injection, patients continued to be stable. Uchida and Suzuki [164] came to the same conclusion in their review of all PET and single photon emission tomography studies of LA injectable antipsychotics. They found that the CNS effects as measured by D2 receptor occupancy of LA injectable antipsychotics can persist for several months, and, as such, these medications may be administered at dosing intervals greater than those recommended in product monographs. The Uchida and Suzuki [164] review included a total of 14 studies with LA injectable FGAs (haloperidol decanoate, n = 11, and fluphenazine decanoate, n = 3), and 4 studies with LA injectable SGAs (LA injectable risperidone, n = 3, and LA injectable olanzapine pamoate, n = 1). Lee et al. [153] reached similar conclusions in their 2015 review of the pharmacokinetic data of LA injectable SGAs available in Canada and the USA (LA injectable aripiprazole, olanzapine, risperidone, and 1- and 3-month formulations of paliperidone palmitate). It should be noted that the clinical studies on intermittent or extended dosing cited here included small numbers of patients. Further clinical studies with larger samples are needed. In addition, future studies must also be designed so that minimal therapeutic dose and intermittent dosing are not confounded, thus allowing a dissociation of effects due to dose versus mode of administration. In summary, continuous occupancy of D2 receptors might not always be necessary to trigger the signaling cascades that underlie the antipsychotic effect. In parallel, the work reviewed here, including preclinical findings [9,11,154], also suggests that sustained antipsychotic treatment above the optimal D2 occupancy threshold for maintenance treatment (which appears lower than for acute and first-episode schizophrenia defined by Kapur [138]) could trigger changes that might be detrimental to the patient. This would include tolerance to therapeutic effects and SP. In parallel, work conducted in animals suggests that intermittent dosing would prevent dopamine supersensitivity [9,11,154]. Both the clinical and animal studies reviewed here suggest that an exploration of intermittent dosing strategies in patients is needed. LA injectable antipsychotics can also be given so as to produce intermittent dosing, taking into account the clinical recommendations of Lee et al. [153] and Uchida and Suzuki [164]. Intermittent dosing involves that D2 occupancy levels be high and then low, with each state maintained for approximately equivalent periods of time. Intermittent dosing should be one way to achieve the minimum therapeutic dose and prevent SP. Thus, LA injectable antipsychotics should not be increased rapidly to mask TD or SP except for palliative treatment in TR patients. However, one should avoid intermittent dosing defined as “targeted” antipsychotic treatment. This involves lengthy intervals without treatment and waiting for prodromal symptoms to reappear before restarting antipsychotic drug treatment. Such approaches were reviewed recently and they turned out to be disastrous for patients [161], since this “targeted” approach led to increased relapse episodes and poor outcomes. Instead, intermittent interval dosing, as used in our animal studies [9,11,154] and as explored in patients by Remington et al. [161], should be investigated further. This would involve intermittent antipsychotic administration with short interdosing intervals (e.g., for oral medication: weekends off and then treatment on Monday to Thursday, as in the McCreadie study [160]). Importantly, the interdosing interval must be long enough to permit peaks and troughs in brain levels of antipsychotic/D2 receptor occupancy. We predict that this enables some level of normal dopamine-mediated neurotransmission to occur in between peaks of D2 receptor blockade by antipsychotics, thus avoiding the neuroadaptations that lead to dopamine supersensitivity [165,166]. The interdosing interval to achieve peaks and troughs in antipsychotic brain levels will be a function of the t1/2 of the antipsychotic. For instance, the length of the interdosing interval would be longer for LA antipsychotics and comparatively shorter for short-acting antipsychotics. Finally, while threshold levels of D2occupancy have already been established for first-episode schizophrenia and acute treatment [138], they must still be established for maintenance treatment.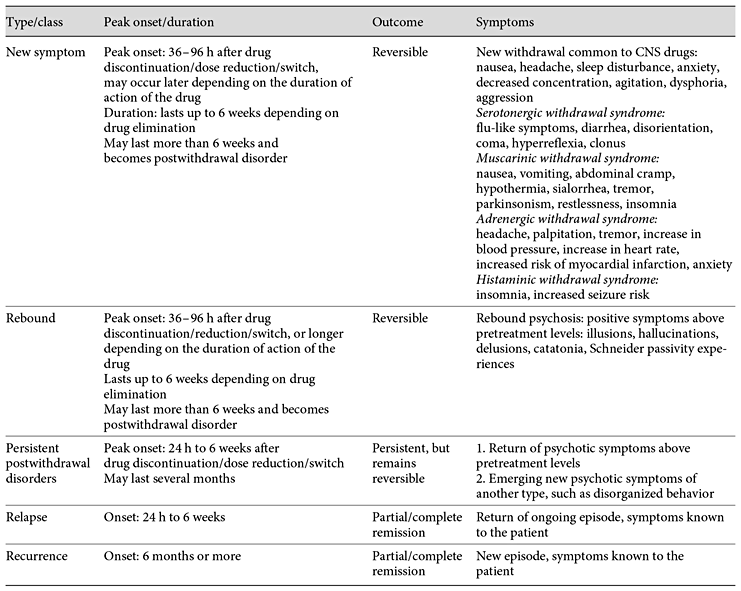

-
Hello . I took anti depressants ,anti anxiety , syzophenia medicines for one month . One doctor prescribed me these medicines after knowing the side effects in inherent I was scared to eat it and didn't want to eat it . I started suffering from acidity after eating I felt scared all the time 24/7 . My legs shivered atoagocally when I tried sitting still , my brain stopped working normally , I stopped eating properly , I stopped caring about everything , then I went to another psychiatrist . He told me I have been prescribed wronge medicines . Which is the syzophenia one cause I don't have syzophenia . I was shocked :((( cause I already ate it for 1 month ... I had to leave school when I promoted to class 9 cause I couldn't concentrate at all it's been a year I threw away those medicines and also complained to the psychiatrist about the side effects . I had brain pain after I stopped taking the 1 mg medicines each . Now I don't have brain pain . every time I tried studying my brain pained I was always a good student first time ever I failed I cried day and nighth I couldn't study and remember things whereas I was the best student layer after seeing my state my parents supported me and complained to psychiatrist . I'm still crying while writing this I thought I will never ever recover and I can't live with brain pain forever . I cried everyday and slowly the brain pain went . I'm healing . Thankgod I didn't eat it for 3 months but I completed 50% course . I think I'll fail next year too . Will I ever recover full my teachers noticed changes in me and told my parents what happened to me . I have heard from 2 people they suffered same things but got addicted to medicines
-
https://www.madinamerica.com/2016/11/frenzy-lobbying-21st-century-cures/ “A Frenzy Of Lobbying On 21st Century Cures” Kaiser Health News and NPR report on the immense lobbying effort aimed at passing the "21st Century Cures" Act which would fast-track FDA approval of drugs and devices. "The 21st Century Cures Act set for a House vote Wednesday is one of the most-lobbied health care bills in recent history, with nearly three lobbyists working for its passage or defeat for every member on Capitol Hill. More than 1,455 lobbyists representing 400 companies, universities and other organizations pushed for or against an earlier House version of a Cures bill this congressional cycle, according to federal disclosure forms compiled by the Center for Responsive Politics. A compromise version was released over the holiday weekend." More at above link, including comments on how to oppose the bill.








